Introduction
There is a growing necessity in neurotoxicology to conduct predictive, high-throughput in vitro screening assays. The National Academy of Sciences has emphasized the need for such assays to identify hazards among thousands of chemicals currently lacking toxicological information.17 Ideally this type of assay would be conducted at the cellular or network level in native cell types to provide sufficient translation for predicting outcomes in vivo. However, existing “phenotypic” screening platforms are poorly suited for the defining phenotype of neurons, the action potential, and its communication between neurons via the synapse. For example, while high content imaging using Ca2+ and voltage-sensitive dyes can detect neuronal activation, important behaviors such as rapid bursting of action potentials in a single neuron can take place at rates higher than video frame capture. On the other hand, high-throughput patch clamp can address rapid bursting of action potentials, but only in dissociated cells, and thus can not detect toxicity at the synapse, or physiological effects on network-level activity. In addition, many insecticides, convulsants, metals and a wide range of natural toxins cause acute effects on the behavior of neuronal networks, which cannot be properly observed with current approaches.3
Based on the these considerations, an ideal in vitro platform for phenotypic screening of neurons and cardiomyocytes would have the following characteristics:
1) Direct recording of the phenotypic signal of interest: voltage,
2) a sample rate sufficient to accurately capture the shape and timing of action potentials,
3) numerous electrodes per well recorded simultaneously to assay synaptic connectivity and cardiac action potential propagation,
4) label-free, non-invasive operation to avoid perturbation of natural cell function, and,
5) preservation of cellular interconnectivity.
While several current screening technologies meet a subset of these criteria, only the microelectrode array (MEA) meets all of them. Neuronal networks on MEAs have shown sensitivity to neurotoxins in many previous studies5,6, and the reproducibility of MEAs in neurotoxicity screening has been demonstrated.16 Unfortunately a limiting factor of previous MEA technology has been the low throughput due to hardware shortcomings. In this application note, we describe the identification of neuroactive and neurotoxic chemicals using the 768-channel MaestroTM multiwell MEA system. A “training set” of chemicals was selected based on previous studies.3 Using simple measures of neuronal activity; high selectivity and sensitivity were observed.
Materials and Methods
Chemicals
See Table 1 for information regarding the sources, purities, and means of delivery for all chemicals used. All chemicals were prepared as 50mM stock solutions in their respective solvents, stored at -20°C in glass vials treated with Sigmacote, and diluted at 1:1000 into the media for a final concentration of 50 µM. The assembled training set consisted of 23 “positive” compounds identified as neuroactive in previous studies (references listed in Table 1), and seven negative control compounds that did not affect neuronal activity in previous studies. A majority of these previous studies employed low-throughput single-well MEA systems. All other chemicals were obtained from commercial vendors and were reagent quality or better.
Multiwell MEA plates
Axion BioSystems’ 12-well microelectrode array plates were used for the electrophysiological data collection. Each well contained an 8 x 8 array of 64 embedded nano-porous platinum electrodes (30µm diameter, 200µm center-to center spacing), for a total of 768 channels. Each well was coated with a 50 µg/mL concentration of poly-L-lysine (Sigma Chemicals, St. Louis, MO). This solution was then removed after 1 hr, and the plates were left to dry at 18°C until the day of culture.
Cell culture on MEAs
Cell culture was conducted using cells from LongEvans rats on postnatal day 0-1. Pups were decapitated, and the neocortex was separated from the brain and placed in a cortical buffer. The soaked cortex was minced finely with scissors and triturated until homogenous. Cells were then filtered through a 100 µm screen. The area of the electrode grid was treated with a 1 mg/mL 50 µL drop of laminin (Sigma) solution. Following dissection, (~45 min), the laminin was removed by vacuum aspiration and 50 µL of media containing 2.5 x 105 cells was placed directly over the electrode grid in each well. The cells were left to adhere for 10-15 minutes before adding 1 mL of Neurobasal-A (NBA) media (500 mL Neurobasal-A; GIBCO), 14 g sucrose (Sigma), 1.25 mL glutamine (200 mM; GIBCO), 5 mL glutamate (2.5 mM, Sigma), 5 mL penicillin/streptomycin (Sigma), and 50 mL FBS (GIBCO), pH 7.4. The day after cell plating (day in vitro 1, or DIV 1), NBA media was replaced with NBA + B27 media (NBA + 10 mL B-27 (GIBCO), pH 7.4). At DIV 4 NBA + B27 media was replaced with glutamate-free NBA + 10%, FBS media. The plates were inspected at least once every 7 days for evidence of evaporation, replacing the lost media as it was necessary.
MEA recording
Spontaneous network activity from cortical cultures was recorded with the Axion multiwell MaestroTM MEA system, consisting of a 768-channel MaestroTM MEA “plate reader”, a MiddleManTM data acquisition interface, and a personal computer running Axion Integrated Studio (AxIS) software. Voltage was sampled at 12.5 kHz on all electrodes simultaneously using a gain of 1200x. Real-time spike detection was conducted using the AxIS adaptive spike detector, a variant of the now established ‘AdaBandFit’ detector method.1 Recordings were conducted at 37°C.
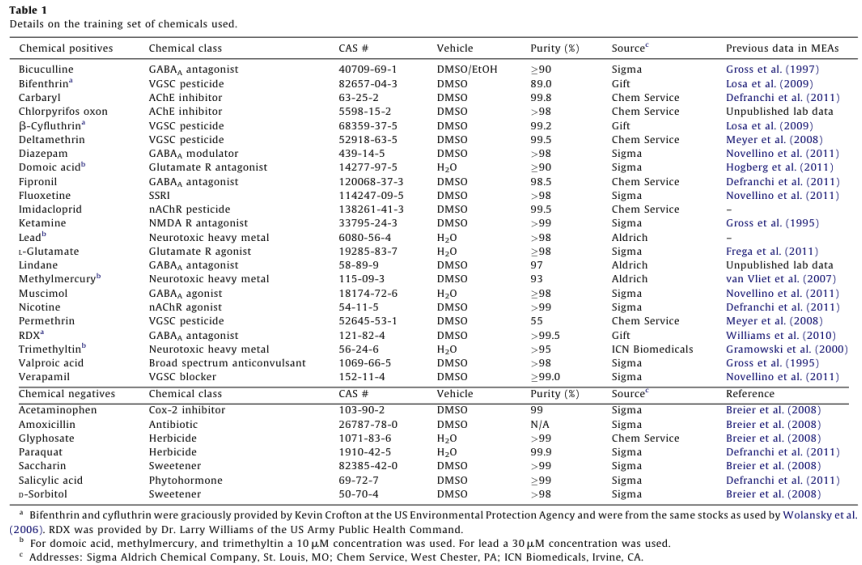
Dosing experiments were conducted between DIV 12 and 22, with a median 14 DIV for all plates. Wells that did not show spontaneous activity on the day of chemical application were not used. Electrodes with rmsnoise levels >10µV were grounded. The experimental protocol for chemical testing is summarized in Fig. 1 and described in detail below.
Raw voltage data were passed through a 1st order 200 - 5000 Hz Butterworth bandpass filter. Spikes were counted in 1s bins using an adaptive threshold set at 8x the standard deviation of the estimate noise for each channel and 33 min of baseline data were collected. Chemicals were applied by diluting 1µL of concentrated stock in 200 µL of media from the appropriate well, and returning the mixture to its well of origin. 33 min of data were collected post-dose. The pattern of chemical application for each plate was randomized (www. randomizer.org). Each plate also contained a vehicle (1µL of dimethylsulfoxide, Sigma) and positive control (50 µM bicuculline methiodide, Sigma). Following dosing with training set chemicals, 1µM tetrodotoxin (TTX) was added to each well, and 10 min of activity was recorded, which served as an assay selective control that decreased activity
Data analysis
Spike count files generated with the AxIS software were used to calculate the number of active electrodes (AEs; defined as an electrode having an average of more than 5 spikes/min) in each well, the average per well mean firing rate (MFR), and the standard deviation of the average per-well MFR. The first 3 min of data were discarded to ensure a stable baseline was achieved. Wells with fewer than 10 active electrodes were not analyzed. Electrodes were also removed post hoc if changes in spike rate were indicative of noise; data from just 4 electrodes from 4 separate wells were removed for this reason.
MFR was first calculated for both the baseline and the treated cases for each electrode, and average per well MFR values were then calculated. To calculate a weighted mean firing rate (wMFR), per-well MFR averages were weighted by the total number of active electrodes in the well using Microsoft EXCEL’s SUMPRODUCT function, and then averaged across a sum of those electrodes. A percent change in wMFR and AE was then calculated for each chemical.
Propidium iodide/Hoechst staining for cytotoxicity
Cytotoxic effects of the 30 chemicals studied were assayed by propidium iodide (PI), which binds to the DNA only in cells with a compromised plasma membrane. Employing the same cell culture technique as illustrated above, 96 well plates were cultured with a cell density of 40,000 cells/well. At DIV 13 or 14, chemicals were added (at concentrations equal to those used in MEA testing) to each well and then incubated at 37°C for 30 min. A positive control was included using triton x100 (1% by volume) in one well per plate to induce cell death. A well in each plate was also reserved as a negative control with no chemical addition. Following the 30 min incubation, the media was removed and 100 µL of 5 µM PI (Invitrogen) in Locke’s buffer was added to each well with wide orifice pipette tips to reduce mechanical disturbance in the cultures. The culture plates were then incubated for another 20 min, after which the PI was aspirated off. 100 µL of a warm fixative containing 2 µg/mL Hoechst 33342 was added to each well and incubated at room temperature for 20 min. The fixative was then aspirated and each well was washed two times with 100 µL of Ca2+/Mg2+-free PBS (Gibco), which was left in the wells after the 2nd wash. These plates were then sealed with optical tape and stored until image collection at 4°C.
A Cellomics ArrayScan VTI (ThermoScientific, Pittsburgh, PA) fluorescence imager was used to count the PI positive cells in each well. Fluorescent images were obtained in two channels: valid cells were identified using Hoescht stain (Hoescht stains DNA in all cell nuclei) by imaging in channel 1 using an excitation wavelength of 365 +/- 25 nm and an emission wavelength of 515 +/- 10 nm. Cells that were PI positive were imaged in channel 2 with the use of an excitation wavelength of 549 +/- 4 nm and an emission wavelength of 600 +/- 12 nm. These images were then analyzed using the Cellomics Target activation Bioapplication. All Hoechst stained nuclei in channel 1 were added to a mask which was used to identify cells in channel 2. When the average fluorescence intensity for a nuclei was above more than 3x the background, nuclei in channel 2 were considered positive. Each well was analyzed to find the final percentage of PI positive cells, and a minimum of 300 cells were counted per well. Each chemical was applied to two wells in two different cultures (n = 4). The values from these four trials were then averaged to determine overall cytotoxicity.
Results
Training set response
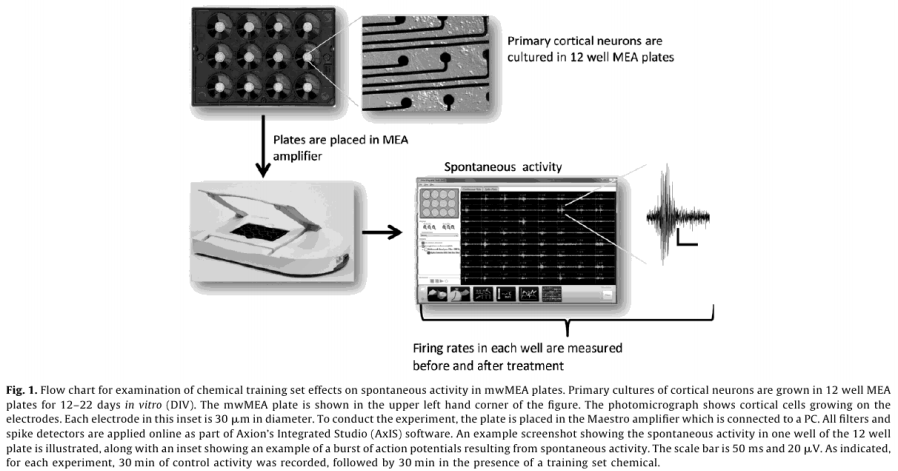
A steady pattern of increased spike activity emerged starting around DIV 5, and reached a steady state around DIV 12, both in terms of the number of active electrodes (AEs), and MFR (Fig. 2).
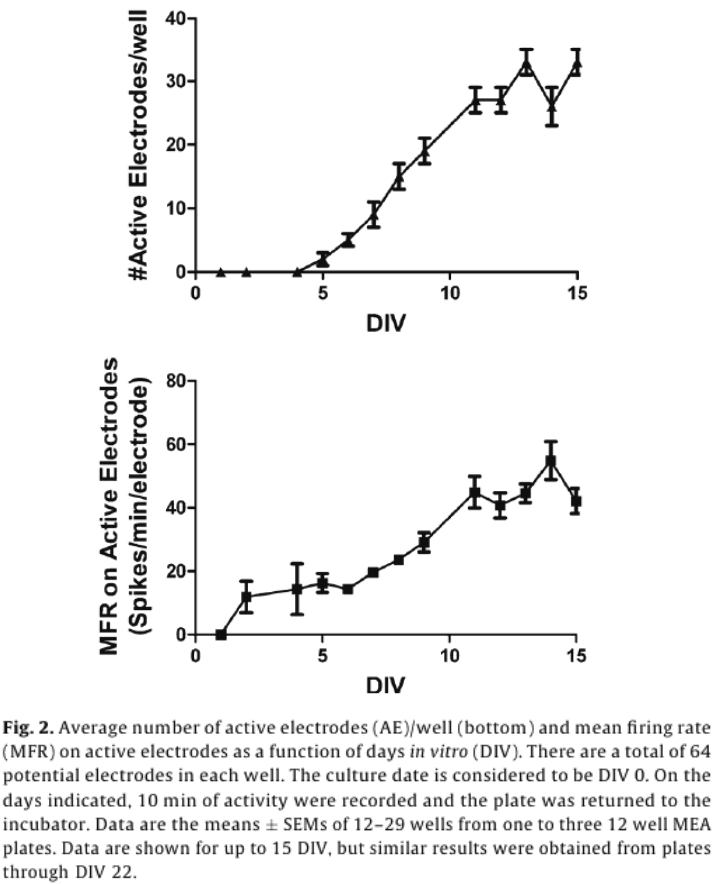
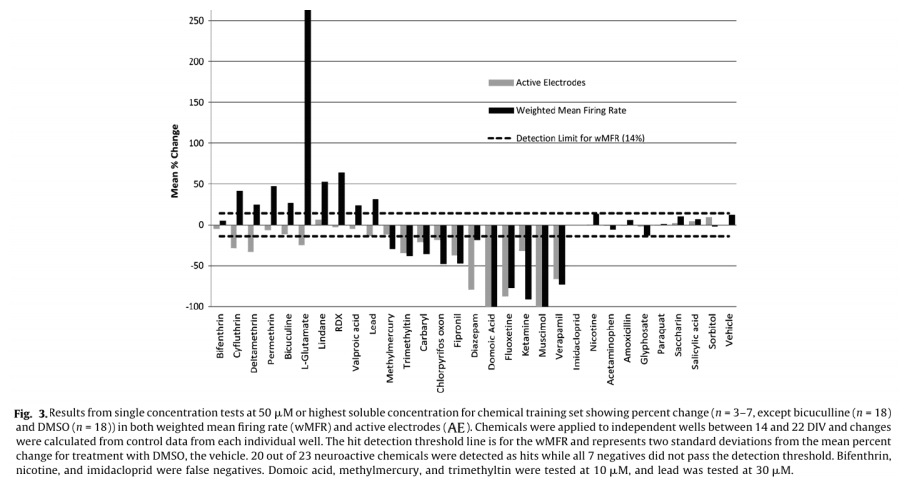
For dosing, each well represented its own control, and changes in activity caused by a chemical treatment were then recorded as a percent of control activity, and used to identify “hits”. A change in MFR is often used as a metric for drug and chemical induced changes in network activity.5,6,14,16,18 To account for the variability in AE in different treatment groups, MFR was weighted by AE (wMFR) for both the control and treated conditions. The threshold for identifying a “hit” was set to 14%, which approximated the mean percent change in wMFR caused by the vehicle control DMSO (12.2%) plus two standard deviations (2 x 0.93%). Because wMFR normalizes for changes in AE percent change, the number of AE was used as a secondary indicator of a “hit”. DMSO did not change the number of active electrodes. To account for biological variability, a 20% or greater change in AE was also considered a “hit”.
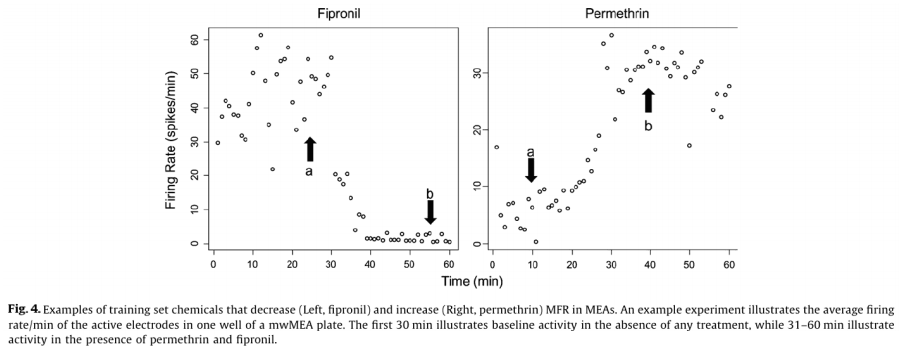
Of the 23 positive compounds listed, 20 caused a change in wMFR that exceeded the threshold values of 14% (Table 2 and Fig. 3). Of these, 12 also caused a change in AE exceeding the threshold of 20%. With few exceptions (cyfluthrin, deltamethrin, and L-glutamate), wMFR and AE typically showed the same directionality. The time course of representative compounds that decrease (fipronil) and increase (permethrin) MFR is illustrated in Fig 4. Of the expected positive chemicals, only nicotine, imidacloprid, and bifenthrin were below threshold. These chemicals will need to be studied further, perhaps to identify potential effects on spike bursting. None of the 7 negative control chemicals reach the threshold values for either parameter. In summary, this multi-well MEA assay displayed a sensitivity of 87% for positives, a specificity of 100% based on negatives, and a combined hit accuracy of 90%.
Confirmation of results
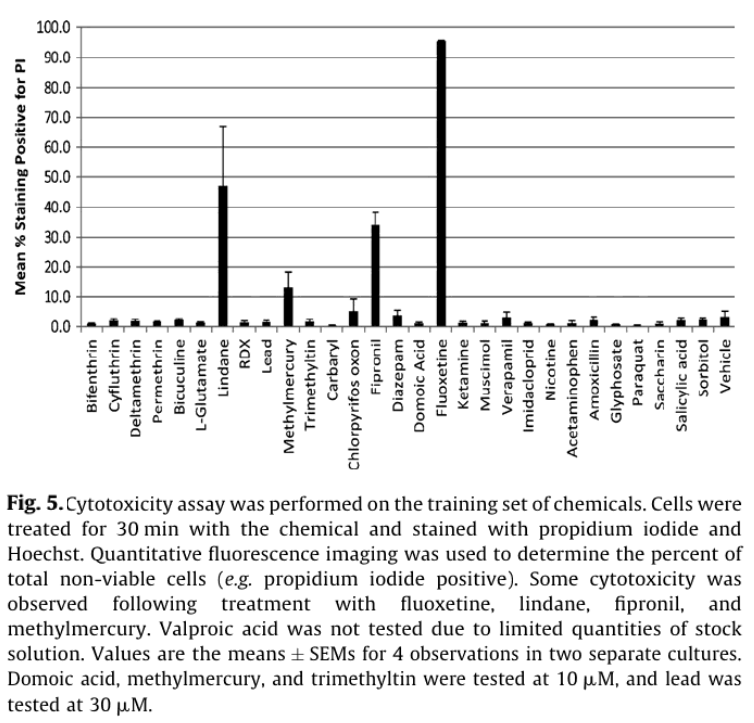
Viability staining was used to distinguish whether the observed electrophysiological effects were driven by cytotoxicity or more direct effects on electrical signaling pathways. Consistent with the latter scenario, only fluoxetine, fipronil, methymercury, and lindane caused >10% cytotoxicity after a 30 min exposure (Fig. 5). Of these fluoxetine (50 µM) caused the most cytotoxicity with 95 +/- 5% of cells staining PI positive (n = 4), consistent with previous studies with glioma and human blastoma cells.13
Discussion
In this application note, we introduce a multiwell microelectrode array format that allows parallel screening of compounds for neuroactivity. Positive chemicals in the training set are known to target a wide variety of receptors (e.g. GABA, glutamate, and cholinergic), channels (NaV) and enzymes (acteylcholinesterase). Using only two simple measures of spike activity (wMFR and AE) and a single dose, neurotoxic chemicals were identified with 87% sensitivity and 100% specificity. While this approach was chosen to maximize screening throughput, higher sensitivity might be achieved by examining chemical effects on spike timing for individual electrodes (e.g. burst rate), or correlation of activity between electrodes in a given well (e.g. synchrony). Only four compounds produced measurable cytotoxicity, indicating that for the majority of the compounds tested, changes in neuroactivity were due to acute effects on cellular electrophysiology. This also highlights the potentially higher sensitivity of neurotoxicity assays based on electrophysiology vs. those with more conventional metabolic or viability endpoints.
wMFR and AE provide little, if any information about molecular mechanisms of action. Where a large set of compounds is being screened using such simple measures, identified hits would typically be followed up with a “2nd tier” assay such as a dose-response experiment. Spike time stamps are easily saved with the MaestroTM system, and analysis of spike timing and correlation of activity between electrodes has been used previously to “fingerprint” compounds based on molecular targets.11 MEA studies such as this would also be complemented by a subsequent target-based screen (e.g. HT patch clamp) for identified hits.
While in vitro assays based on biochemical or morphological endpoints can be conducted at relatively high throughput, it can be difficult to predict how changes in these endpoints will affect neuronal function. While electrophysiological approaches directly address the functional output of neurons, existing platforms have, thus far, lacked the throughput necessary for efficient screening. Axion’s multiwell MaestroTM MEA platform offers a significant increase in the capacity to measure compound-induced alterations in neuronal network function. The SLAS-compliant design of the multiwell MEA plate will facilitate future incorporation into multimodal systems employing automated plate handling.
References
1. Biffi E, Ghezzi D, Pedrocchi A, Ferrigno G. Development and Validation of a spike detection and classification algorithm aimed at implementation on hardware devices. Comput Intell Neurosci
2010;2010:1–15.
2. Breier J, Radio N, Mundy WR, Shafer TJ. Development of a high-throughput assay for assessing anti-proliferative compounds using human neuroprogenitor cells. Toxicol Sci 2008;105:119–33.
3. Crofton K, Mundy W, Lein P, Bal-Price A, Coecke S, Seiler A, et al. Developmental neurotoxicity testing: recommendations for developing alternative methods for the screening and prioritization of chemicals. Altern Anim Exp 2010;28(1):9–15.
4. Defranchi E, Novellino A, Whelan M, Vogel S, Ramirez T, van Ravenzwaay B, et al. Feasibility assessment of mirco-electrode chip assay as a method of detecting neurotoxicity in vitro. Front Neuroeng 2011;4(6):112.
5. Frega M, Pasquale V, Tedesco M, Marcoli M, Contestabile A, Nanni M, et al. Cortical cultures coupled to micro-electrode arrays: a novel approach to perform in vitro excitotoxicity testing. Neurotoxicol Teratol (August)2011 [Epub ahead of print].
6. Gopal K. Neurotoxic effects of mercury on auditory cortex networks growing on microelectrode arrays: a preliminary analysis. Neurotoxicol Teratol 2003;25: 69–76.
7. Gramowski A, Schiffmann D, Gross GW. Quantification of acute neurotoxic effects of trimethyltin using neuronal networks cultures on microelectrode arrays. Neuro-toxicology 2000;21:331–42.
8. Gross GW, Rhoades BK, Azzazy HME, Wu M-C. The use of neuronal networks on multielectrode arrays as biosensors. Biosens Bioelectron 1995;10:553–67.
9. Gross GW, Harsch A, Rhoades BK, Gopel W. Oder, drug and toxin analysis with neuronal networks in vitro: extracellular array recording of network responses. Biosens Bioelectron 1997;12(5):373–93.
10. Hogberg HT, Sobanski T, Novellino A, Whelan M, Weiss DG, Bal-Price AK. Application of micro-electrode arrays (MEAs) as an emerging technology for developmental neurotoxicity: evaluation of domoic acid-induced effects in primary cultures of rat cortical neurons. Neurotoxicology 2011;32:158–68.
11. Johnstone AF, Gross GW, Weiss DG, Schroeder OH, Gramowski A, Shafer TJ. Microelectrode arrays: a physiologically based neurotoxicity testing platform for the 21st century. NeuroToxicology 2010;31(4):331–50.
12. Keefer EW, Norton SJ, Boyle NAJ, Talesa V, Gross GW. Acute toxicity screening of novel AChE inhibitors using neuronal networks on microelectrode arrays. NeuroToxicology 2001;22:3–12.
13. Levkovitz Y, Gil-Ad I, Zeldich E, Dayag M, Weizman A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines. J MolNeurosci 2005;27:29–42.
14. Losa SM, Johnstone AFM, Shafer TJ. Relative potencies of type I and type II pyrethroids for inhibition of spontaneous firing in neuronal networks. Toxicol CD Soc Toxicol 2009;108(1) Abstract # 2126.
15. Meyer DA, Carter JM, Johnstone AFM, Shafer TJ. Pyrethroid modulation of spontaneous neuronal excitability and neurotransmission in hippocampal neurons in culture. Neurotoxicology 2008;29:213–25.
16. Novellino A, Scelfo B, Palosaari T, Price A, Sobanski T, Shafer T, et al. Development of micro-electrode array based tests for neurotoxicity: assessment of interlaboratory reproducibility with neuroactive chemicals. Front Neuroeng 2011;4(4):1–14.
17. NRC. Toxicity testing in the twenty-first century: a vision and a strategy. Washington, DC: The National Academies Press; 2007.
18. Schroeder OH-U, Gramowski A, Jugelt K, Teichmann C, Weiss D. Spike train data analysis of substance-specific network activity: application to functional screening in pre-clinical development. In: Proceedings of the 6th international meeting on substrate-integrated microelectrodes. July 8–11, 2008, Reutlingen, Germany; 2008.
19. van Vliet E, Stoppini L, Balestrino M, Eskes C, Griesinger C, Sobanski T, Whelen M, Hartung T, Coecke S. Electrophysiological recording of re-aggregating brain cell cultures on multi-electrode arrays to detect acute neurotoxic effects. Neurotoxicology 2007;28:1136–46.
20. Williams LR, Johnstone AFM, Bannon DI, Shafer TJ. RDX binds to the convulsant site of the GABAA receptor and increases spontaneous firing rates of cortical neurons Toxicol CD Soc Toxicol 2010;114(1) Abstract # 216
21. Wolansky MJ, Gennings C, Crofton KM. Relative potencies for acute effects of pyrethroids on motor function in rats. Toxicol Sci 2006;89:271–7.



