Key Findings
>> MEA reveals reduced neuronal activity in homozygous ALS disease model cells
Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects both cortical and spinal motor neurons. Examination of neurons from ALS patients reveals the existence of atypical protein aggregates in the cells’ cytoplasm. The specific proteins found in these aggregates can vary, but approximately 95% of ALS patients develop aggregates containing the TDP-43 protein (transactivation DNA-binding protein 43)1. In rare genetic forms of ALS, this can be caused by mutations such as M337V in the TDP-43 (TARDBP) gene2. Despite significant research and funding, the development of therapies for ALS has been challenging due to a lack of readily accessible human cell models, leading scientists to rely on animal models and cell lines that differ considerably from human biology.
In this Application Note, Charles River Laboratories (Cambridge, UK) functionally characterized ioGlutamatergic Neurons TDP-43 M337V/ WT™, ioGlutamatergic Neurons TDP-43 M337V/M337V™ and wildtype ioGlutamatergic Neurons™ developed by bit.bio (Cambridge, UK). Together, these cells form a genetically matched disease model for the investigation of ALS in vitro. For this purpose, they used the Axion Maestro Pro multiwell microelectrode array (MEA) platform to assess electrophysiological differences between the disease model cells and the wild-type control, observing decreased neural activity in the homozygous disease model.
Introduction
ALS is a progressively fatal neurodegenerative disease that damages nerve cells in the central nervous system (CNS), but its effects are manifested in the peripheral nervous system (PNS) due to the loss of motor neuron function and the resulting muscle weakness and atrophy. Although ~85-90% of ALS cases are sporadic, mutations in over 40 genes have been identified that contribute to the development of ALS, including in the TARDBP gene, which encodes TDP-43. Mutations in TARDBP are also associated with frontotemporal dementia (FTD) and, in both disorders, cause increased phosphorylation, mislocalization, protein aggregation, and altered mRNA splicing of genes such as stathmin-2 (STMN-2).
Despite considerable research efforts and funding, there is no cure for ALS, and approved treatments are primarily aimed at addressing symptoms. A significant hurdle in early stage drug discovery for ALS is the lack of standardized, easy to use, and readily accessible human cell models. Instead, scientists have been constrained by animal models and cell lines that differ considerably from human disease biology, posing a considerable challenge for lead optimization.
Human induced pluripotent stem cell (iPSC)-derived disease models offer an advantage over traditional in vitro models by providing a more accurate representation of clinically relevant disease phenotypes. However, the process of differentiating iPSCs to the desired cell type using directed differentiation suffers from inherent variability as result of stochastic events, complex protocols, and external signaling cues. This can lead to heterogeneous populations and difficulties in connecting genotype to phenotype, and ultimately hinders scale up for high throughput applications in drug discovery. Furthermore, an important consideration for iPSC-based disease modeling is the need for proper controls to mitigate the influence of genetic background on disease manifestation and progression3. bit.bio’s precision cell reprogramming technology, opti- ox™, aims to resolve these inconsistencies and inefficiencies to enable the precise, consistent, and scalable production of human cells. This technology has been used to develop a range of iPSC-derived ioGlutamatergic Neurons carrying disease-specific mutations. Within 11 days, human stem cells convert consistently into defined ioGlutamatergic Neurons using a straightforward, two-phase protocol, accessible to scientists without stem-cell expertise (Fig. 1). Using the parental glutamatergic neuron cell line, disease models were developed by engineering a heterozygous or homozygous M337V mutation into the TARDBP gene, resulting in ioGlutamatergic Neurons TDP-43 M337V/WT and ioGlutamatergic Neurons TDP-43 M337V/ M337V. Both disease models can be used alongside the genetically matched (isogenic) wild-type control ioGlutamatergic Neurons in phenotypic and screening assays for early-stage drug discovery and the development of new therapeutics for ALS.
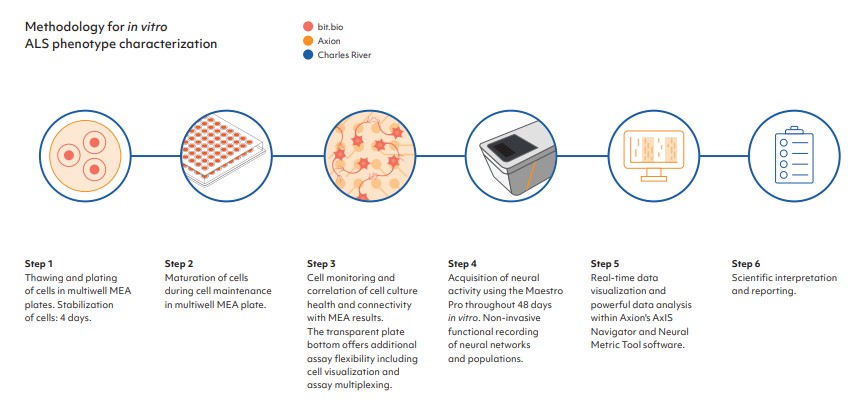
Functional electrophysiological measurement is important for the characterization of neurodegenerative disease phenotypes and to identify differences between a disease model and isogenic control. Axion BioSystems’ Maestro Pro MEA platform provides label-free assessment of neural activity and connectivity over time, allowing for simple analysis of complex biology. The Maestro Pro detects key aspects of neural networks, including activity, synchrony, and oscillation, to provide a comprehensive functional profile for disease modeling. Standard format multiwell plates allow for high throughput evaluation of disease models and isogenic control cells, and their response to potential therapeutics.
The work presented in this Application Note was performed at Charles River Laboratories by a multidisciplinary scientific team with extensive expertise in discovering and characterizing novel drug candidates for ALS. Charles River Laboratories offers a comprehensive portfolio of in vitro models and cell-based assays to screen, identify and validate new drug candidates. By combining physiologically relevant cells with high content imaging, phenotypic assays and biomarker readouts, Charles River supports ALS drug development, for both small molecules and advanced therapies, across hit-to-lead screening processes, lead optimization and in vitro pharmacology.
Results
Wild-type and disease model ioGlutamatergic Neurons develop structural neuronal networks at comparable rates
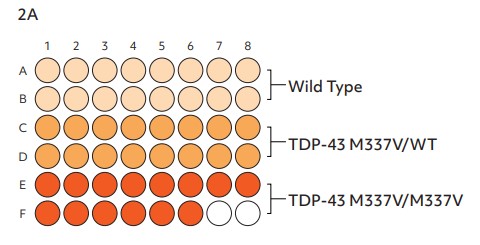
To verify the cells as suitable disease models for functional screening in lead optimization workflows, it is critical to identify whether the genetically engineered disease models demonstrate a disease relevant phenotype when compared to the isogenic control. The wild-type and disease model glutamatergic neurons were co-cultured with human iPSC-derived astrocytes at a ratio of 5:1 in a single plate (Fig. 2A) and their functional neuronal electrical activity was assessed at several time points on the environmentally controlled Maestro Pro MEA platform. Both wild-type and disease model cells developed a stable monolayer in the plates, with suitable coverage of the electrodes.
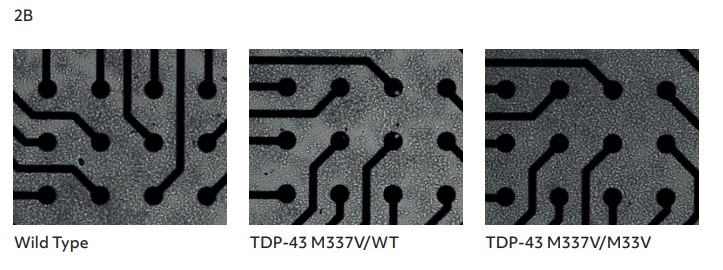
Brightfield imaging at 26 days in vitro (DIV) showed the expected neuronal morphology of neurite outgrowth and the formation of structural neuronal networks (Fig. 2B), enabling high quality recording of electrical activity.
MEA reveals reduced neuronal activity in homozygous ALS disease model cells
The firing activity of the cells was assessed twice per week from DIV 7 to 46, by 5-minute MEA recordings, to identify when synchronous firing was established. Data were recorded using the ‘Neural Spontaneous’ configuration in AxIS Navigator software (Axion BioSystems). Action potentials were detected via adaptive thresholding to assess weighted mean firing rate and network burst frequency.
Weighted mean firing rate (Fig. 3A, 3C) and network bursting (Fig. 3B, 3C) increase over time in culture to produce synchronous network firing of neurons, which was quantifiable from DIV 34. ioGlutamatergic Neurons TDP-43 M337V/M337V show a clear reduction in both firing rate and network burst frequency when compared to the ioGlutamatergic Neurons TDP-43 M337V/WT and the wild-type control (Fig. 3A, 3B).
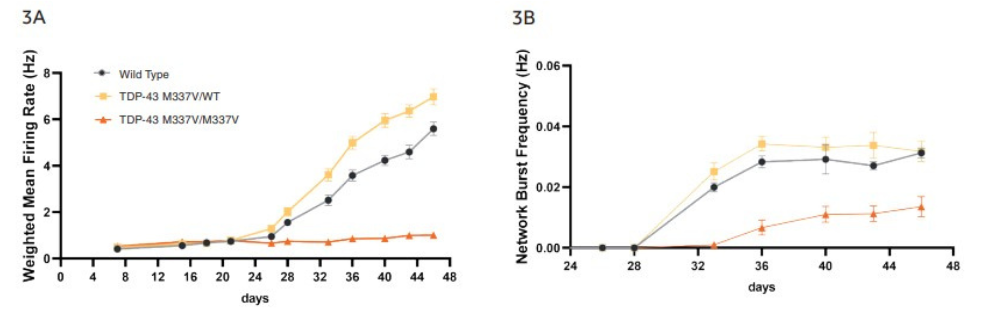
Quantification of raster plots over the course of culture shows that ioGlutamatergic Neurons TDP-43 M337V/M337V have a lower weighted mean firing rate (Fig. 3A) and network burst frequency (Fig. 3B) than wild-type and ioGlutamatergic Neurons TDP-43 M337V/WT. No clear difference is noted between wild-type and TDP-43 M337V/WT. Error bars indicate SEM, n=8-15 technical repeats. Neurons produce clear burst and network burst activity as seen in the raster plots at DIV 46 (Fig. 3C). In the raster plots, each dash indicates a firing event; blue indicates a single electrode burst; and the pink box indicates a network burst event.

Discussion
The Discovery team at Charles River Laboratories used Axion’s MEA platform to compare network activity and functional network formation of bit.bio’s wildtype ioGlutamatergic Neurons and isogenic disease model cells carrying the heterozygous or homozygous mutation M337V in the TDP-43 gene, associated with ALS and FTD.
The Maestro Pro MEA platform offers the advantage of transparent, multiwell plates, enabling simultaneous characterization of disease model and wild-type control cells in similar conditions. This also enables simultaneous brightfield imaging for confirmation of cell spotting accuracy and correlation of cell culture health and connectivity with MEA results.
The results showed a clear reduction in the neuronal activity and network formation of the ioGlutamatergic Neurons TDP-43 M337V/M337V disease model cells, compared to the wild type and heterozygous disease model cells, indicating their potential as a physiologically relevant translational model for ALS and FTD.
This robust electrophysiological assay developed by the Charles River Discovery team can be offered to clients alongside high content imaging and gene expression analysis to investigate TDP-43 protein phosphorylation, mislocalization, aggregation, and altered mRNA splicing, which are hallmarks of ALS and FTD. This comprehensive portfolio of cell-based assays can add significant value at the hit-to-lead and lead optimization phases of a preclinical workflow to advance drug discovery and development for these devastating neurodegenerative diseases.
References
[1.] Glass, Jonathan D. (2020). Stathmin-2: Adding Another Piece to the Puzzle of TDP-43 Proteinopathies and Neurodegeneration. J. Clin Invest, 130(11), 5677-5680.
[2.] Scotter, E.L., Chen, H.J., Shaw, C.E. (2015). TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics, 12(2), 352-63. Erratum in: Neurotherapeutics, 2015, 12(2):515-8.
[3.] Guo, W., Fumagalli, L., Prior, R., and Van Den Bosch, L. (2017). Current Advances and Limitations in Modeling ALS/FTD in a Dish Using Induced Pluripotent Stem Cells. Front Neurosci. 11, 671.
Authors
Charles River, bit.bio, and Axion BioSystems


